Amino Acid Metabolism
Humans ingest more protein (amino acids) than they need for replacement of endogenous proteins. These excess amino acids can not be stored and thus are catabolized (metabolized).
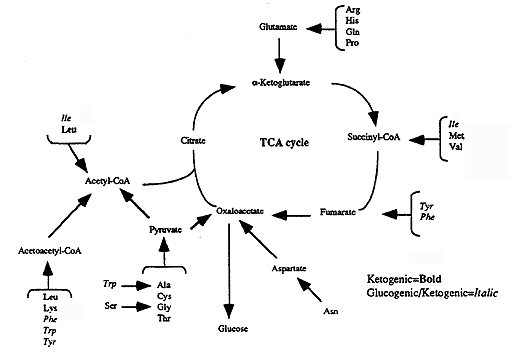
Ultimate Fate of the carbon from excess amino acids: CO2, and energy (ATP) via TCA cycle and respiratory chain
Metabolic Fates of Amino Acids:
Ketogenic vs. Glucogenic:
Glucogenic- amino acids which can be converted into glucose (CHO producing), Pyruvate or a TCA cycle intermediate that can be converted to OAA is produced in the final step of its metabolism.
Ketogenic- amino acids which can be converted into fat (fat producing), Acetyl CoA or Acetoacetyl CoA is produced in the final step of their metabolism. (Acetyl CoA condenses with OAA so the net gain of OAA is zero since one molecule is consumed for each molecule of Acetyl CoA produced)
| Glucogenic | Ketogenic | Glucogenic & Ketogenic |
| Ala, Gly | Leu | Ile |
| Arg, His | Lys | Phe |
| Asn, Met | Trp | |
| Asp, Pro | Tyr | |
| Cys, Ser | ||
| Gln, Thr | ||
| Glu, Val |
Metabolism of Phe and Tyr (Liver):
Demonstrates a number of important features common to Glucogenic and Ketogenic pathways:
(1) production of organic intermediates
(2) requirements for co-factors
(3) the possibility of inborn errors of metabolism at multiple steps
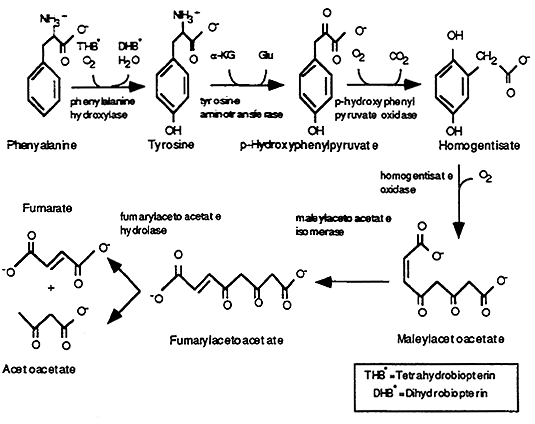
[1] Phenylalanine Hydroxylase: Phe -----> Tyr
Requires: oxygen
Co-factor: Tetrahydrobiopterin, synthesized by animals and other microorganisms.
Deficiencies: Phenylketonuria (PKU, common inborn error in metabolism), results in elevations of Phe, which if very high, prolonged --> permanent neurologic damage
Solution: dietary restriction of Phe, all newborns screened to detect PKU --> low Phe diet
Tetrahydrobiopterin Deficiencies: defects in synthesis can also result in elevations of blood Phe and severe neurologic disorders.
[2] Tyrosine Aminotransferase: Tyr -----> p-Hydroxyphenylpyruvate
Deficiencies: elevated blood Tyr (but not Phe since it is an irreversible reaction) ---> Tyr crystals form in tissues (cornea, palms, soles)
Solution: restrict dietary Tyr
[3] p-Hydroxyphenylpyruvate Oxidase: p-Hydroxyphenylpyruvate -----> Homogentisate
Co-factor: Ascorbic Acid
[4] Homogentisate Oxidase: Homogentisate ------> Maleylacetoacetate
Requires: Oxygen
[5] Maleylacetoacetate Isomerase: Maleylacetoacetate ----> Fumarylacetoacetate
Deficiency: severe "back-up" of the pathway producing very high levels of Phe and Tyr, usually occurs w/in the first months of life often requiring liver transplantation
Metabolism of Met and Cys:
Features:
(1) pathway for excretion of sulfur generated from the sulfur containing amino acids: Met and Cys
(2) co-factors: cobalamin and folic acid
(3) important "methyl" group generator for synthesis of a number of cmpds.
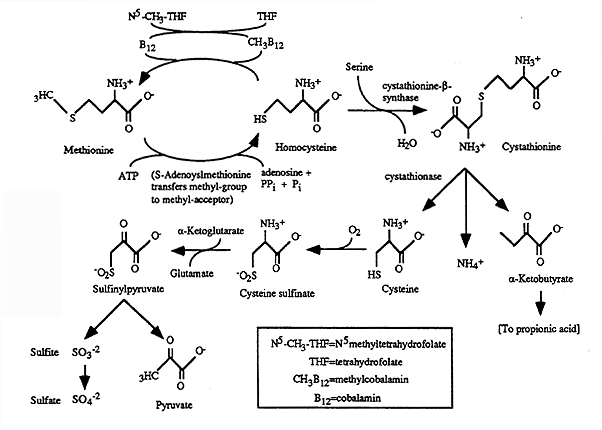
[1] Methione Adenosyltransferase:
Met ----> Homocysteine
Transfer of Methyl onto adenosine resulting in S-Adenosylmethione (SAM)
SAM- "methyl" donor during synthesis of creatine, phosphatidylcholine, epinephrine etc.
Homocysteine -----> Met: cobalamin (B12) containing enzyme, methylcobalamin donates a "methyl" group to homocysteine.
[2] Cystathione-b-synthase: Homocysteine ----> Cystathione
Co-factor: pyridoxine
Deficiency: homocystinuria, increased levels of homocysteine which has a reactive sulfur group that can bind to a number of proteins.
[3] Cystationase: Cystathione ----> Cysteine
Co-Factors- Review
Co-Factors: Organic and Inorganic
Organic: Prosthetic Groups (i.e. Heme) and Co-enzymes (derived from water soluble vitamins)
Review of Water Soluble Vitamins
Pyridoxine (B6): Used in many rxns, particularly those involving transamination. Conversion of homocysteine to cystathione.
Cobalamin (B12): Used in conversion of homocysteine to methione.
Folic Acid: Used in the synthesis of folates important in "methyl" group transfers.
Vitamin C (Ascorbic Acid): Used in many reactions, primarily as a reducing agent.
© Dr. Noel Sturm 2019