Positive and negative ions in an aqueous solution, otherwise soluble with the counter ions in their environment, produce highly insoluble precipitates with certain added reagents.
Chapter 3, Gravimetry
Gravimetric methods of analysis are used where weights of reactants and products of chemical reactions are reproducible, stable and reflect the presence of constituents which are important in the establishment of identity.
Two important methods deal with the trapping and weighing of products in the solid and
gaseous phases. The
first of these falls into the category of a precipitation method.
3-1. Precipitation methods. Many metallic elements in their ionic forms react with negative
counter ions to
produce stable precipitates. Silver ions form stable and highly insoluble salts with chloride,
bromide and iodide.
Calcium precipitates quantitatively with oxalate and can be measured reproducibly at any of three
temperature
dependent plateaus as the oxalate, the carbonate and the oxide. Barium precipitates
quantitatively
as the sulfate.
The reactions often follow the same patterns:
![]()
![]()
Positive and negative ions in
an aqueous solution, otherwise soluble with the counter ions in their environment, produce
highly
insoluble precipitates with certain added reagents.
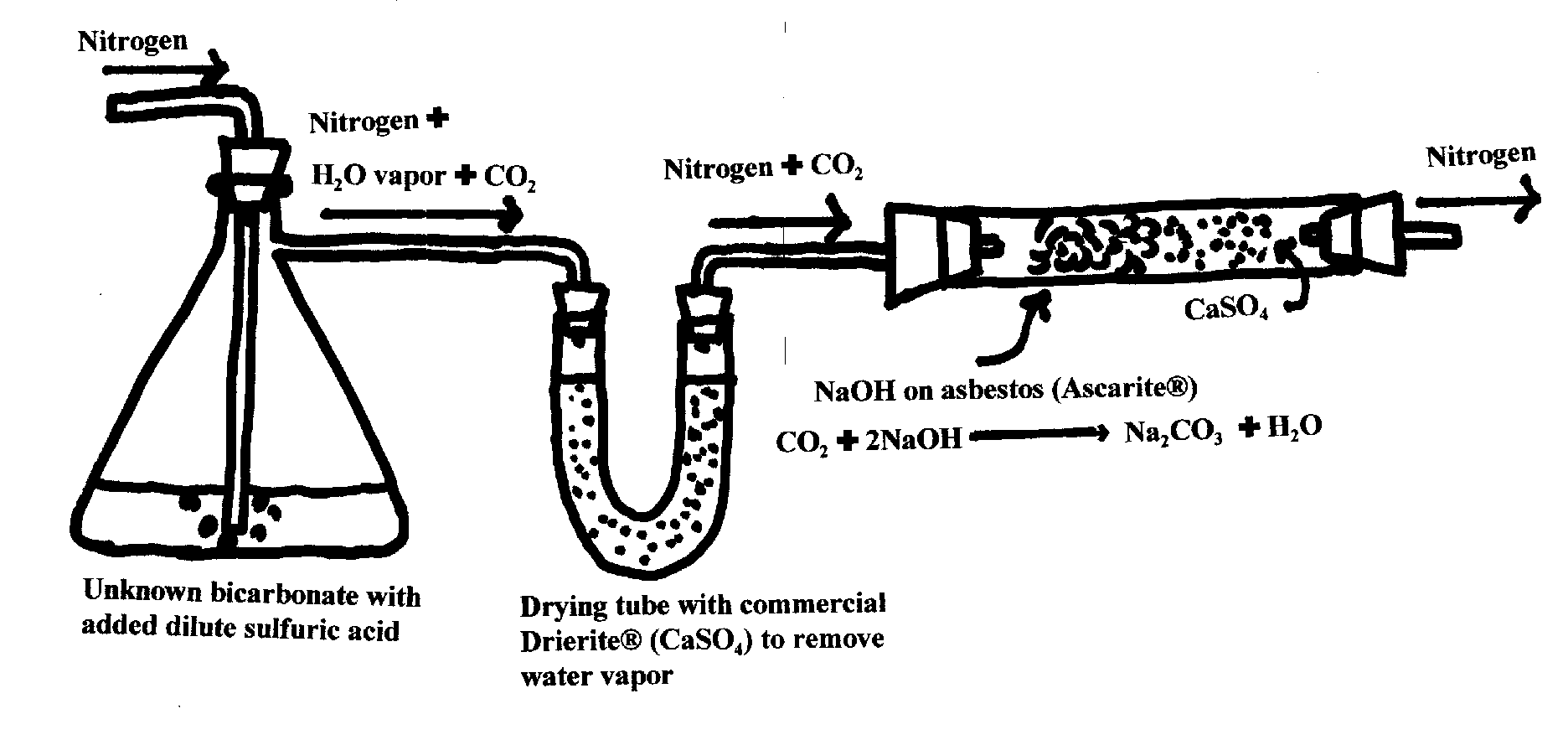
3-2. Volatilization methods. An interesting volatilization method which is entirely
gravimetric
is the one shown
by the equations below.
The analyte can be bicarbonate as shown or a mixture of carbonate and bicarbonate. The
total amount of
carbonate in whatever form is found by placing the analyte in a solution containing an excess of
H2SO4. This
solution is in a flask connected to incoming nitrogen gas gently bubbled through the solution and
an exit tube
first to a drying agent to absorb aerosolized water and water vapor and then to a mixture of
NaOH and drying
agent to absorb the CO2 and ![]()
water subsequently produced by the absorption by NaOH:
![]()
The apparatus is
shown below. The tube containing the NaOH on asbestos and the CaSO4 to absorb
the final
water product is pre- and post-weighed to given the total amount of carbonate in the sample.
Note that the
nitrogen gas acts only as a carrier and does not take part in any reaction.
3-3. Considerations for the isolation of precipitates
Precipitates ought to be easy to wash free of contaminants without loss of the precipitate
either in solution or
through the filter. The particle size of the precipitate ought to be large enough not to escape
through the filter
pores. That the precipitate has a low solubility is paramount. The precipitate ought not to react
with the
atmosphere and it must have a known composition which remains stable after ignition.
3-3a. Substances of low solubility have the nasty habit of forming colloidal suspensions.
Colloidal particles have
diameters from 10-7 cm to 10-4 cm. That is on the order of from 10
atomic diameters to 10,000 atomic diameters.
Particles in this size range are still sufficiently jostled about by thermal molecular motion to
remain in suspension.
Where they are the result of a process of precipitation brought about by the addition of ionic
species, the
particles are surrounded by the excess ionic species. If Ba2+ is added in excess to
SO42- the BaSO4 precipitate which is formed is
considered to be surrounded by Ba2+ ions. If the opposite procedure were being
followed, the
precipitate would be surrounded by SO42- ions. That these particles
all have like charge and therefore repel each other suggests that your technique must favor the
formation of large rather than small precipitate particles and to offer ways
of encouraging
the coagulation of particles after they have formed.
This can be done by carrying out the precipitation at a temperature close to the boiling point
of water, in a dilute
solution of your analyte and with constant stirring for the reasons given below.
Although analytical chemists still have some disagreement as to the mechanism of precipitation, there is wide agreement that a quantity called the relative supersaturation affects the particle size. Relative supersaturation is given as
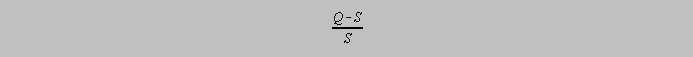
where Q is the
instantaneous concentration of the species added to effect precipitation and S is the equilibrium
solubility of the substance which precipitates. Particle size seems to be inversely proportional to
Relative
Supersaturation because a high concentration of added reagent increases the probability that
oppositely charged
ions will begin the precipitation process at late as well as early stages of the addition and the
resulting particles
will be on the order of atomic dimensions, whereas the maintenance of a value of Q just slightly
above S lowers
that probability but offers in any case a layer of the added reagent ions around existing particles
for their further
growth.
3-4. The Electric Double Layer
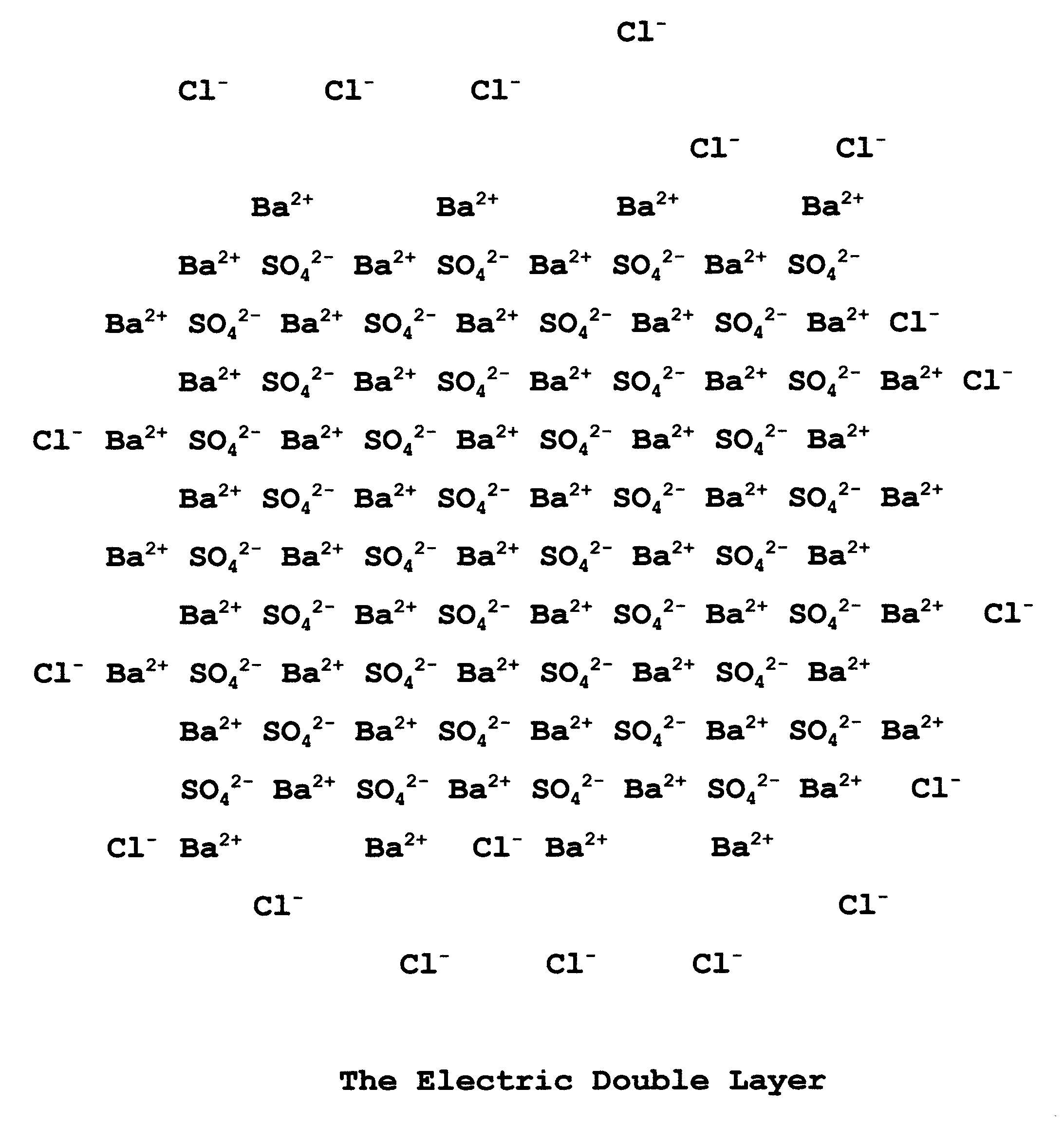
If a particle of precipitate is surrounded by the ion in excess, say Ba2+ in the case of the determination of SO42-, any negative ions in the immediate surroundings will be attracted to that primary positive layer. In the case of the addition of a BaCl2 solution to a Na2SO4 solution, the ions Cl- and SO42- are available. As the sulfate is used up in the precipitation process it is the chloride which is left and which forms the second layer. Thus we have an electric double layer, made up first of barium ions then of chloride ions. This double layer keeps the colloidal precipitate particles from coming into contact with each other for further coagulation.
There are two ways to bring the
particles closer together and to
increase the probability of
coagulation: (1) heating increases
overall thermal motion, affecting both
the mobility of adsorbed ions and of
the colloidal precipitate particles
themselves. The summary effect is
that there are collisions of particles
which result in the increase in particle size due to increased coagulation; (2) increasing the
electrolyte concentration
of the solution, for reasons not entirely clear, results in a decrease in the mean radius of the
electric double layer and
encourages further coagulation. Carrying out both of these operations results in
digestion of the precipitate, an unfortunate term because
biological
digestion usually refers to the dissolving of food and absorption at the molecular
level through the wall of the intestine. Digestion in quantitative
analysis refers to the coagulation of a precipitate
into a filterable form. Unfortunately after successful digestion, some of the primary electric layer
is made up of Na+
ions which must be washed away ultimately for quantitative results to be achieved. The
Ba2+ ions as well will give
a positive error if not removed and end up being dried with the precipitate as excess
BaCl2. Many coagulated
precipitates do not respond well to washing with distilled water because as the second electric
layer is removed
(excess Cl- for example) the first remains on all particles with an electric charge of
the same sign. The result is that
there is a return to the repulsive state and an effective increase in the radius of the particles which
then begin once
again to separate as colloidal particles. The process is called
peptization and is to be avoided if some of the precipitate is not to
be lost. One way around this for many precipitates is to encourage digestion by heating and
also by increasing the electrolyte concentration by washing with a reagent which will go off as a
gas during the drying
process. Dilute nitric acid, HNO3 , is effective for washing excess ions from AgCl.
In choosing such a wash, it is
imperative that the procedure has been carried out and has been
shown to yield
reproducible, quantitative results. Unexpected side reactions, complex formation and changes in
solubility with
added reagents are sufficiently unpredictable to make intuition in the absence of experience
unacceptable.
3-5. Other demons which can plague quantitative precipitate isolation.
During the precipitation procedure a number of other problems can arise to give erroneous
positive or negative
results. Among these are surface adsorption, mixed crystal formation, occlusion and mechanical
entrapment.
Any ions may be carried down during a precipitation as the result of surface
adsorption. Na+ , or Cl- in the case of the
determination of SO42- by the addition of dilute BaCl2
solution to a NaSO4 solution. Both Ba2+ and Na+ can
compete for lattice positions as the particles form. Likewise, the ions Cl- and
SO42- can have the same effect. In the quantitative determination of
some transition metals, iron for example as Fe(OH)3, zinc, cadmium and
manganese
may be present as impurities and all three form sparingly soluble hydroxides as well, though each
with greater
solubility than the hydroxide of iron:
| Compound | Solubility Product |
| Fe(OH)3 | 4 x 10-38 |
| Cd(OH)2 | 2.5 x 10-14 |
| Mn(OH)2 | 1.9 x 10-9 |
| Zn(OH)2 | 1.2 x 10-17 |
Mixed crystal formation can occur if two ions have the same
charge, if their ionic diameters are sufficiently close
to fit into the same crystal lattice. Ions which commonly interfere with each other are shown in
the table below with
their ionic diameters in picometers given after each.
| Interfering | Ions |
| K+, 133 pm | NH4+, 148 pm |
| Sr2+, 113 pm | Ba2+, 135 pm |
| Mn2+, 80 pm | Cd2+, 97 pm |
In cases where one has a known interference of one ion with the other it is necessary to find
methods of removing
one before carrying out a precipitation of the other, or using a precipitating reagent in which there
is no interference.
Occlusion and mechanical entrapment. If a precipitation
procedure is carried out too quickly, pockets of solvent
and spectator ions can form, trapping them within the precipitate particles and dashing one's hope
of removing them
during the washing procedure. This is another reason why the relative supersaturation must be
kept as low as
possible so that in principle at least, all precipitation occurs only at the surface of a growing solid
particle, devoid
of solvent pockets.
All of these problems of coprecipitation of unwanted ions can lead to positive or negative
errors. In the example
above where it pointed out that Na+ or Cl- may coprecipitate in the
SO42- determination, surface adsorption will produce a positive error.
In the case of
mixed crystal formation, the direction of the error depends on the relative atomic weight of the
ion which replaces that which is desired in the precipitate. In the case of the precipitation of zinc
hydroxide, mixed
crystal formation with manganese would produce a negative error but with cadmium or zinc a
positive error.
| Desired precipitate | |||
| Compound | Mn(OH)2 | Zn(OH)2 | Cd(OH)2 |
| At. Wt. of M2+ | 54.94 | 65.39 | 112.41 |
| Direction of error | negative | --- | positive |
3-6. The use of the technique of homogeneous solutions to effect precipitation.
A solution containing a reagent which produces a desired ion to effect precipitation, often by
gentle heating of the
solution, offers an exquisite means for obtaining well-formed large crystal particles which lend
themselves splendidly
to the technique of filtration.
The model we use to explain why this happens also uses the concept of relative
supersaturation. The initial
nucleation of sparingly soluble particles offers a surface template
which favors "locking" onto ions in the vicinity
which by the luck of the draw (and the kinetic molecular theory) find themselves at the right
energy and orientation
to enter the crystal lattice. Ions isolated from a growing crystal are not favored to enter this
process because at least
two are required, both at the right energy and orientation to start the growth of a new crystal. If
the concentration of one ion of a sparingly soluble salt increases gradually by slow homogeneous
synthesis in a solution, then as its concentration reaches the threshold of supersaturation for the
ion pair, a relatively small number of nucleated particles grows to larger size (because the
probability of finding a place in an existing crystal lattice for any single ion is greater than that
of
a spontaneous creation a new crystal from dissolved and randomly arranged ions) rather than a
large number of nucleated particles growing in constant competition with the rest and thus
remaining small. The result for the latter is a non-filterable precipitate, but one in the former
which filters
quite well.
See the demonstration of this effect at
http://155.135.31.26/oliver/demos/prechomo/prechomo.htm
Here is a table of common reagents useful for the preparation of ions often needed for
precipitation processes.
| Reagent | Precipitating species | Precipitation reaction | Elements which yield to
this reaction |
| Urea | OH- | (NH2)2CO + 3H2O --->
CO2 + 2NH4+ + 2OH- | Al, Ga, Th, Bi, Fe, Sn |
| Trimethyl phosphate | PO43- | (CH3O)3PO + 3H2O ---> 3CH3OH + H3PO4 | Zr, Hf |
| Ethyl oxalate | C2O42- | (C2H5)2C2O4 + 2H2O ---> 2C2H5OH + H2C2O4 | Mg, Zn, Ca |
| Dimethyl sulfate | SO42- | (CH3O)2SO2 + 4H2O ---> 2CH3OH +SO42- + 2H3O+ | Ba, Ca, Sr, Pb |
| Trichloroacetic acid | CO32- | Cl3CCOOH + 2OH- ---> CHCl3 + CO32- + H2O | La, Ba, Ra |
| Thioacetamide | H2S | CH3CSNH2 + H2O --->
CH3CONH2 + H2S |
Sb, Mo, Cu, Cd |
| Dimethyl glyoxime | CH3(CNOH)2CH3 | CH3COCOCH3 +
2H2NOH ---> DMG + 2H2O |
Ni |
| 8-Acetoxyquinoline | C9H6NOH | CH3COOQ + H2O ---> CH3COOH + HOQ | Al, U, Mg, Zn |
3-7. Preparation of a dry weight of your precipitate.
The resulting precipitate must be heated until a stable dry state is reached. Some
understanding of typical precipitate
properties is mandatory for repeatable results to be achieved.
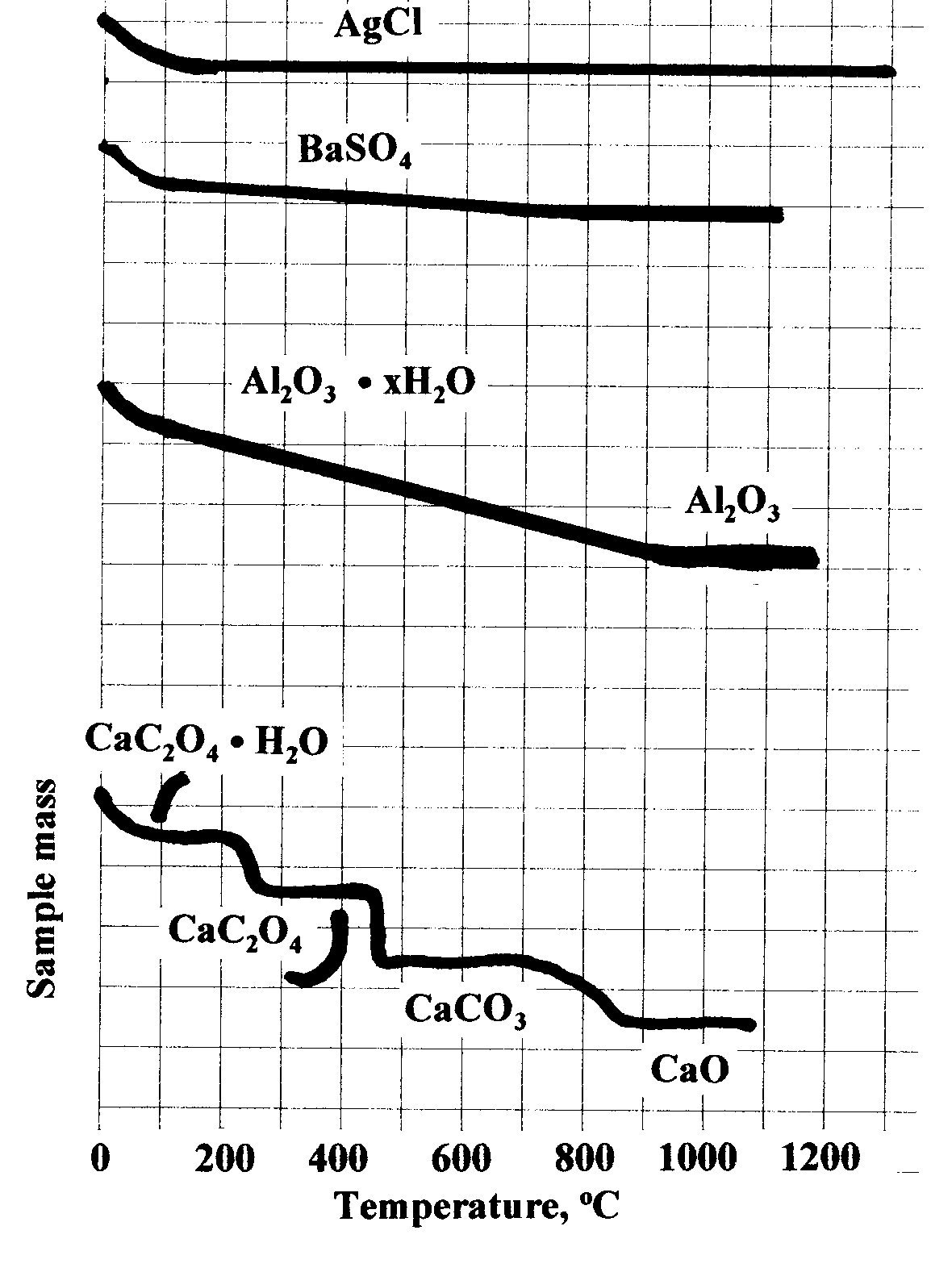 Note in the figure at the
right that whereas AgCl
achieves a stable dry weight just above 100oC, BaSO4 does not do so
until it reaches a temperature in the
vicinity of 700oC Aluminum oxide, Al2O3, loses water
slowly as the temperature rises to 1000oC at which point it achieves stability. Some
compounds
decompose in several stages, reaching stable plateaus.
Calcium oxalate, CaC2O4 H2O, loses all its water at
around 200oC and remains stable as CaC2O4 until just
above 400oC at which point it decomposes to calcium carbonate,
CaCO3 where it remains stable up to
700oC. Between 700oC and 850oC it slowly
decomposes to CaO where it remains stable until its
melting point at 2614oC.
Note in the figure at the
right that whereas AgCl
achieves a stable dry weight just above 100oC, BaSO4 does not do so
until it reaches a temperature in the
vicinity of 700oC Aluminum oxide, Al2O3, loses water
slowly as the temperature rises to 1000oC at which point it achieves stability. Some
compounds
decompose in several stages, reaching stable plateaus.
Calcium oxalate, CaC2O4 H2O, loses all its water at
around 200oC and remains stable as CaC2O4 until just
above 400oC at which point it decomposes to calcium carbonate,
CaCO3 where it remains stable up to
700oC. Between 700oC and 850oC it slowly
decomposes to CaO where it remains stable until its
melting point at 2614oC.
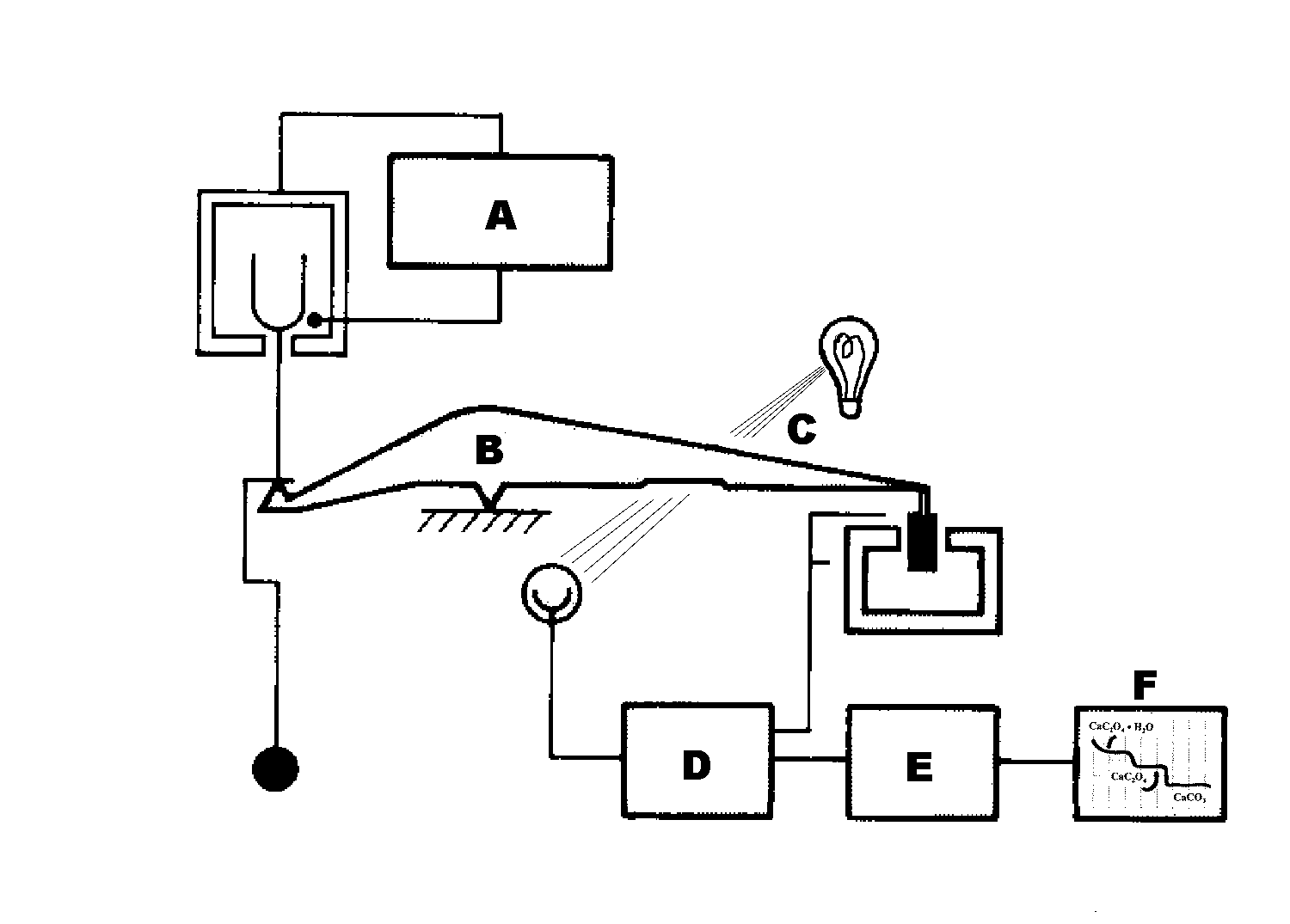
A device not seen often in analytical laboratories but
useful for producing automatic plots of mass of
sample vs. temperature such as those at the right is
the thermobalance (below). Region A includes the heating circuit, a
temperature sensor, sample cup and
counter weight resting on one end of the balance arm,
B. A light wave is partially attenuated at C, giving a
negative feedback to the amplifier circuit at D,
designed to yield an output voltage which increases
with the force necessary to keep the balance in
equilibrium (and the attenuation at a constant value).
One can adjust the baseline voltage at E, the tare adjuster, so as to produce the graph at the chart
recorder, F.
Example 3-1: A 0.3427 g sample of bronze-age jewelry
is analyzed for silver content by first dissolving it in
concentrated nitric acid and precipitating it as AgCl. The precipitate is transferred to a dry
sintered glass filter
weighing 12.2347 g where it is separated from the filtrate and washed with dilute nitric acid. The
filter and
precipitate are dried at 150oC, cooled, weighed and found to weigh 12.4373 g.
Calculate the %Ag in the jewelry.
 (To be solved and
discussed in class)
(To be solved and
discussed in class)
Example 3-2. A sample of iron ore weighing 0.4275 g is dissolved in 12M HCl. The
resulting
solution is slowly
made basic with NaOH until the first hint of a turbid
solution is detected. 10.0 g urea are dissolved and the
solution heated just to the boiling point for 4 hours. The precipitate,
Fe2O3 x H2O, is trapped using ashless filter paper. The
precipitate and filter paper are fired in a porcelain crucible of empty weight 12.2837g until the
ashless
filter paper is completely incinerated and anhydrous Fe2O3 is left.
The
resulting weight of crucible and precipitate is 12.4274 g. Determine the
%Fe, the %Fe2O3 and the %Fe3O4.
(To be solved and discussed in class)
Example 3-3. A sample known to contain only KCl and NaCl and weighing 0.4263 g is
dissolved in water and
treated to an excess of AgNO3, using standard methods of precipitation. The AgCl
precipitate is caught on a Gooch
Crucible of original dry weight of 15.2748 g. The AgCl precipitate is dried at
150oC, cooled and the crucible and precipitate are found to weigh
16.2872 g. Determine the %KCl and the %NaCl in this sample.
Solution is based on the difference in the %Cl in the pure salts:
| Salt | %Cl |
| NaCl | 60.66 |
| KCl | 47.55 |
(To be solved and discussed in class)
3-8. Preferred methods of gravimetric analysis. Most inorganic ions have yielded to
gravimetric analytical
techniques, but one finds many interfering ions. The table below illustrates both the abundance
of
reagents available
for use as well as the problems which can be encountered by interfering ions:
| Analyte | Precipitate | Measured form | Interferences |
| K+ | KB(C6H5)4 | KB(C6H5)4 | NH4+,Ag+,Hg2+, Tl+,Rb+,Cs+ |
| Mg2+ | Mg(NH4)PO4.6H2O | Mg2P2O7 | Many metals (none from Na+ and K+) |
| Ca2+ | CaC2O4.H2O | CaCO3 or CaO | Many metals (none from Mg2+, Na+ and K+) |
| Ba2+ | BaSO4 | BaSO4 | Na+,K+,Li+,Ca2+,Al3 + ,Cr3+,Fe3+,Sr2+,Pb2+ |
| Ti4+ | TiO(5,7-dibromo-8-hydroxyquinoline)2 | TiO(5,7-dibromo-8-hydroxyquinoline)2 | Fe3+,Zr4+,Cu2+,C2O4 2-, citrate, HF |
| VO43- | Hg3VO4 | V2O5 | Cl-,Br-,I-,SO42- , CrO42-,AsO43-,PO43-< / sup> |
| Cr3+ | PbCrO4 | PbCrO4 | NH4+,Ag+ |
| Mn2+ | Mn(NH4)PO4.H2O | Mn2P2O7 | Interferences from numerous metals |
| Fe3+ | Fe(HCO2)3 | Fe2O3 | Interferences from numerous metals |
| Co2+ | Co(1-nitroso-2-naphtholate)3 | CoSO4 (by reaction with H2SO4 ) | Fe3+,Zr4+,Pd2+ |
| Ni2+ | Ni(dimethylglyoximate)2 | Ni(dimethylglyoximate)2 | Pd2+,Pt2+,Bi3+,Au3+ |
| Cu2+ | CuSCN | CuSCN | NH4+,Pb2+,Hg2+,Ag+ |
| Zn2+ | Zn(NH4)PO4.H2O | Zn2P2O7 | Interferences from numerous metals |
| Ce4+ | Ce(IO3)4 | CeO2 | Th4+,Ti4+,Zr4+ |
| Al3+ | Al(8-hydroxyquinolate)3 | Al(8-hydroxyquinolate)3 | Interferences from numerous metals |
| Sn4+ | Sn(cupferron)4 | SnO2 | Cu2+,Pb2+,As(III) |
| Pb2+ | PbSO4 | PbSO4 | Ca2+,Sr2+,Ba2+,Hg2+, Ag+,HCl, HNO3 |
| NH4+ | NH4B(C6H5)4 | NH4B(C6H5)4 | K+, Rb+, Cs+ |
| Cl- | AgCl | AgCl | Br-, I-, SCN-, S2-, S2O32-, CN- |
| Br- | AgBr | AgBr | Cl-, I-, SCN-, S2-, S2O32-, CN- |
| I- | AgI | AgI | Br-, Cl-, SCN-, S2-, S2O32-, CN- |
| SCN- | CuSCN | CuSCN | NH4+,Pb2+,Hg2+,Ag+ |
| CN- | AgCN | AgCN | Cl-, Br-, I- , SCN-, S2-, S2O32- |
| F- | (C6H5)3SnF | (C6H5)3SnF | Except
alkali metals, many interferences,
and SiO44- , CO32- |
| ClO4- | KClO4 | KClO4 | |
| SO42- | BaSO4 | BaSO4 | Na+,K+,Li+,Ca2+,Al3 + ,Cr3+,Fe3+,Sr2+,Pb2+ |
| PO43- | Mg(NH4)PO4.6H2O | Mg2P2O7 | Many interferences except Na+,K+ |
| NO3- | Nitron nitrate | Nitron nitrate | ClO4-, I-, SCN-, CrO42-,ClO3-, NO2-, Br-, C2O42- |
| CO32- | CO2 (by addition of acid) | CO2 | CO2 is trapped as Na2CO3 on Ascarite |
There are a number of organic functional groups which precipitate with metal ions by one of
two routes: (1) chelating
agents are organic compounds which "wrap around" a metal ion thanks to cationic side chains
which form coordinate
covalent bonds with the ion, and (2) a straightforward ion-ion bond which produces a new
species
that excludes
water of solvation and thus precipitates. Good examples of chelating agents include Ethylene
Diamine Tetraacetic
Acid (EDTA), oxalic acid, glycine, 8-hydroxyquinoline and dimethylglyoxime.
Some common organic precipitating agents:
| Compound | Ions precipitated |
| Dimethylglyoxime | Ni2+,Pd2+,Pt2+ |
| EDTA (Ethylenediamine tetraacetic acid) | Zn2+, Cu2+, Pb2+, Ca2+, Ni2+, Fe3+ |
| Cupferron | Fe3+,VO2+,Ti4+, Zr4+,Ce4+,Ga3+,Sn4+ |
| 8-Hydroxyquinoline | Fe3+,Al3+,Mg2+,Zn2+, Cu2+,Cd2+,Pb2+, Bi3+, Ga3+,Th4+, Zr4+, TiO2+, UO22+ |
| Salicylaldoxime | Bi3+,Ni2+,Pd2+,Zn2+, Cu2+,Pb2+ |
| 1-Nitroso-2-naphthol | Fe3+,Co2+,Pd2+, Zr4+ |
| Nitron (C20H16N4) | NO3-, ClO4-, BF4-, WO42- |
| Sodium tetraphenylborate | NH4+, organic ammonium, Ag+, Cs+, Rb+, K+ |
| Tetraphenylarsonium chloride | Cr2O72-, MnO4-, ReO4-, MoO42-,WO42-, ClO4- |